A 6m banner hangs down a high, wood-paneled wall in the lofty entrance of the Sanger Institute in Cambridgeshire, eastern England. With its delicate bands of pink, gray and green, it looks like a enormous, abstract watercolor. Closer inspection reveals its true nature, however. Those pale stripes are made up of streams of different colored letters.
More than a million — 1,346,000 to be precise — are printed on the banner. And not just any old letters. Only As, Cs, Gs and Ts, each representing one of the chemical bases of DNA, the stuff of our genes, are inscribed there. This is no giant “watercolor,” but a printout of part of an X-chromosome, one of the packets of genetic material that lie curled within our cells and which direct chemical operations inside our bodies.
However, it is the size, not the content, of the banner that makes it so remarkable — for it turns out those letters represent only a small section of an X-chromosome. A read-out of a whole one would require a further 114 banners, each crammed with more than a million letters. And that is just the start. To display the letters that make up all 23 pairs of chromosomes in the human genome would require a staggering 2,226 banners. The institute would look like a wallpaper factory during a clearance sale.
And that is why the Sanger banner is so important. It vividly demonstrates the human genome’s extraordinary complexity and puts into perspective the remarkable effort that went into its unraveling. That task was completed in June 10 years ago when the genome’s first rough draft was published. This success was hailed as one of the greatest achievements of modern science, a point that was emphasized this month when US biologist Craig Venter revealed that he had assembled an entire computer-generated synthetic genome — not of a human but of a synthetic “bug” — and inserted it into bacteria which had then begun to replicate. Geneticists were playing at God, claimed newspaper headlines.
This is an exaggeration, but Venter’s success does demonstrate how much genetics has changed in the past two decades. This is no longer a lab-bench science that relies on test tubes and pipettes to study living organisms. Instead, it has become utterly dependent on the swelling power of the microprocessor. Look at that banner of genetic data. It could not have been generated without the staggering capacity of modern computers.
The Sanger Institute employs more than 800 scientists and is devoted to the study of biology. Yet its banks of computers now rival those built by Cern in Switzerland to analyze results from the myriad particle collisions produced by its Large Hadron Collider. Each sequencing machine at the center generates strips of letters from small pieces of DNA and this data is then processed by computers to produce a read-out of a full genome — of a human, or an animal like a dog or a bacterium.
It took almost a decade of processing DNA this way in the 1990s to help to produce the 3 billion letters that made up the first sequenced human genome. By 2008, the institute was processing data at a rate of 100 million letters a day, roughly a genome a month, says Phil Butcher, head of IT at the Sanger. “Today, we can handle so much data that we are producing a couple of genomes every 24 hours,” he says. “And, of course, we are making improvements all the time.”
So vast is the institute’s array of computers that it is planning to build its own 2.5-megawatt power station, a combined heat and power unit, which will generate electricity for them. Biology has become as reliant on computer power as hard sciences like particle physics or astronomy. It is an astonishing transformation. The question is: How did it happen? More to the point, what has it achieved so far and what is it likely to achieve in the near future?
Chronic myeloid leukemia is a cancer of white blood cells that usually occurs in the middle-aged and elderly. It is triggered by a genetic mutation that interferes with chemical messages that help to control cell division, leading to the uncontrolled growth of white blood cells. In the past, prognosis was poor — survival periods of around five months were typical. However, prospects for patients with chronic myeloid leukemia have changed dramatically in recent years, thanks to doctors’ new understanding of the human genome, according to Dr David Adams, a Sanger Institute geneticist and cancer expert. “The drug that has changed everything is called Gleevec,” he says, “and it was derived from our new, computer-driven understanding of the genome.”
By studying a key section of the human genome, scientists realized that a mutation there produces a specific protein (called “bcr-abl”) that in turn triggers a cascade of chemical reactions in a patient’s body that results in chronic myeloid leukemia. Awareness of the protein’s role allowed scientists to develop a drug that could block its activity and so halt the proliferation of white blood cells.
“Patients who have the specific mutation that causes chronic myeloid leukemia will respond to the use of Gleevec and will go into remission quite profoundly,” says Adams. “It was understanding the specific genetics of this disease that led to the realization this drug could help.”
It is an encouraging tale that has since been repeated for several other genome-driven anti-cancer drugs, although it is important to note, says Adams, that the success of these drugs is hit and miss — sometimes they produce no effect. But when they do have an impact, it is invariably profound.
Medical impact
This has important consequences, he argues. In the next 10 years, once computing power has reached the stage when it will become possible to provide full read-outs, easily and cheaply, of everyone’s genome, doctors will be able to determine exactly who will benefit from specific cancer drugs and who will not. Similarly, other types of medicine will have their efficacy judged in advance. “If you had the full genome sequence of everyone, you would know exactly who will respond to a drug and who will not. It will be of enormous benefit,” says Adams.
That goal, although distant, does reveal the importance of scientists’ current obsession with decoding not just a single genome but of generating thousands of different ones, a task that now absorbs a host of follow-on projects, including the Cancer Genome Project, the 1,000 Genomes Project and others; these require the constant running of the institute’s huge rooms of computers. By pinpointing changes in a few base pairs possessed by some individuals and not by others, scientists can discover why the former group might be prone to a particular disease but not the latter. Another example is provided by Crohn’s disease, an extremely painful inflammatory disease of the intestines whose origins have, until recently, defied the attention of scientists.
“In the last two years, genome-wide studies at several centers have pinpointed around 30 genes that have variants involved in Crohn’s disease,” says Nicole Soranzo, who works in the gleaming Sanger labs on the genetics of complex diseases. “This is important because these genes reveal the pathways that lead to Crohn’s and are now allowing drug companies to test their different drugs in order to find one that could block that pathway.”
If nothing else, these examples show that the sequencing of the human genome is already having a medical impact, particularly in the case of cancer treatment but that the real improvements still remain out of a reach, a point acknowledged by Sir Mark Walport, director of the Wellcome Trust, which funds the Sanger Institute. “At the start, there was a tendency to say the project would solve all of humankind’s evils. However, it has taken longer than everyone expected so there has been a backlash,” Walport says. “The reality is somewhere in the middle. No, we cannot yet read our own genomes, but we are discovering networks of genes that influence people’s tendencies to develop diabetes, multiple sclerosis and common obesity, which we all hope will be turned into new therapeutic opportunities.”
This will not be an easy task. Pick any two individuals at random and you will find 99.9 percent of their DNA is identical. “Two genomes typically differ by one base in 1,000 or around 3 million bases in total,” says Sanger scientist Chris Tyler-Smith. That arithmetic means that if you want to pinpoint where an A base is substituted for a G in a gene, making a person prone to diabetes or obesity, then hundreds of genomes will have to be compared, each one made up of billions of letters.
Only staggering computing power will provide that delicate, elusive information. Most scientists believe this goal can be achieved though there is a danger, says John Sulston, the Nobel prize-winner and former head of the Sanger Institute, that researchers will get lost in the technology and data-crunching. “At the end of the day, we need to keep a perspective on what we do and need to think about the biology involved in our work. Computers are just the means to an end. We should not forget that.”
This point is acknowledged by scientists, although they remain confident of success. “Yes, looking for a couple of bases among billions is daunting, especially when you are dealing with hundreds of genomes,” adds Walport. “This is a huge informatics challenge but we are dealing with it. We should look at this as a fantastic, mind-boggling phase of scientific discovery.”



President William Lai (賴清德) yesterday delivered an address marking the first anniversary of his presidency. In the speech, Lai affirmed Taiwan’s global role in technology, trade and security. He announced economic and national security initiatives, and emphasized democratic values and cross-party cooperation. The following is the full text of his speech: Yesterday, outside of Beida Elementary School in New Taipei City’s Sanxia District (三峽), there was a major traffic accident that, sadly, claimed several lives and resulted in multiple injuries. The Executive Yuan immediately formed a task force, and last night I personally visited the victims in hospital. Central government agencies and the

Australia’s ABC last week published a piece on the recall campaign. The article emphasized the divisions in Taiwanese society and blamed the recall for worsening them. It quotes a supporter of the Taiwan People’s Party (TPP) as saying “I’m 43 years old, born and raised here, and I’ve never seen the country this divided in my entire life.” Apparently, as an adult, she slept through the post-election violence in 2000 and 2004 by the Chinese Nationalist Party (KMT), the veiled coup threats by the military when Chen Shui-bian (陳水扁) became president, the 2006 Red Shirt protests against him ginned up by
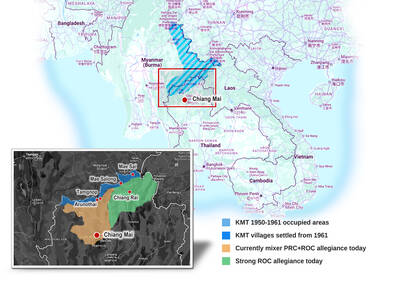
As with most of northern Thailand’s Chinese Nationalist Party (KMT) settlements, the village of Arunothai was only given a Thai name once the Thai government began in the 1970s to assert control over the border region and initiate a decades-long process of political integration. The village’s original name, bestowed by its Yunnanese founders when they first settled the valley in the late 1960s, was a Chinese name, Dagudi (大谷地), which literally translates as “a place for threshing rice.” At that time, these village founders did not know how permanent their settlement would be. Most of Arunothai’s first generation were soldiers

Among Thailand’s Chinese Nationalist Party (KMT) villages, a certain rivalry exists between Arunothai, the largest of these villages, and Mae Salong, which is currently the most prosperous. Historically, the rivalry stems from a split in KMT military factions in the early 1960s, which divided command and opium territories after Chiang Kai-shek (蔣介石) cut off open support in 1961 due to international pressure (see part two, “The KMT opium lords of the Golden Triangle,” on May 20). But today this rivalry manifests as a different kind of split, with Arunothai leading a pro-China faction and Mae Salong staunchly aligned to Taiwan.